Menu
Menu
Genomic research has come a long way since the draft of the human genome was first published over two decades ago, driven by the continuous advancements in next-generation sequencing (NGS) technologies. One of the most transformative methods to emerge from these developments is Hi-C, a library preparation technique that provides unique insights into the three-dimensional organization of the genome.
However, analyzing Hi-C data presents its own set of challenges, particularly regarding data normalization. We’ve created a comprehensive guide to delve into the intricacies of Hi-C data normalization providing you with a deep understanding of its importance and the various tools and methods available for this critical step in the analysis pipeline (see link below).
Normalization approaches for Hi-C data can be categorized into two main types: explicit and implicit. Explicit approaches aim to directly account for individual biases, including GC content, fragmentation, mappability, and enzyme cut sites. They rely on the assumption that these biases are well understood and can be accurately accounted for. The two primary explicit normalization methods are:
Implicit approaches, on the other hand, make use of the assumption of “equal loci visibility.” They assume that cumulative bias is captured within the sequencing depth of each bin of the contact matrix. Some common implicit methods include:
The choice between explicit and implicit methods depends on the level of understanding of biases and the complexity of the genome being studied. Explicit methods require more user-defined parameters, making them suitable for less-studied organisms where biases may be poorly understood. Implicit methods, with their simplicity, are often preferred for well-studied genomes like human and mouse.
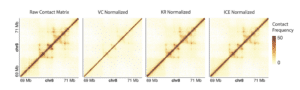
Visual Comparison of Normalization Results.Each contact matrix depicts the same 2Mb region on chromosome 8 of a Micro-C library that was sequenced with 800 million read pairs. The matrix was subjected to different normalization approaches and plotted in R. The scale bar is consistently maintained across each normalization approach to better visualize the impact of normalization. This image clearly demonstrates the challenges associated with using coverage alone as a normalization strategy, whereas the more iterative approaches yield a clearer picture of chromatin interactions.
As the field of Hi-C data analysis continues to evolve, researchers often grapple with the choice of normalization method and pipeline. To date, no single “gold standard” method has emerged. Various studies have compared different algorithms, with Rao et al. opting for the KR method due to its computational efficiency. However, KR may falter with sparse contact matrices, in which case ICE, a robust balancing method, can be used. Overall, SCN, KR, and ICE strategies tend to perform similarly, with only minor differences at lower resolutions.
In practical terms, the choice between these approaches often depends on the tools and pipelines you are using, as many provide KR and ICE as common normalization methods. The field is rapidly advancing, and newer algorithms like Binless may influence the consensus in the future.
For a more detailed explanation of the various methods of data normalization for Hi-C data, we’ve created a white paper. Access it here.